Accurate Haplotype-Resolved Assembly in Human Trios Using Long Fragment Read Sequencing
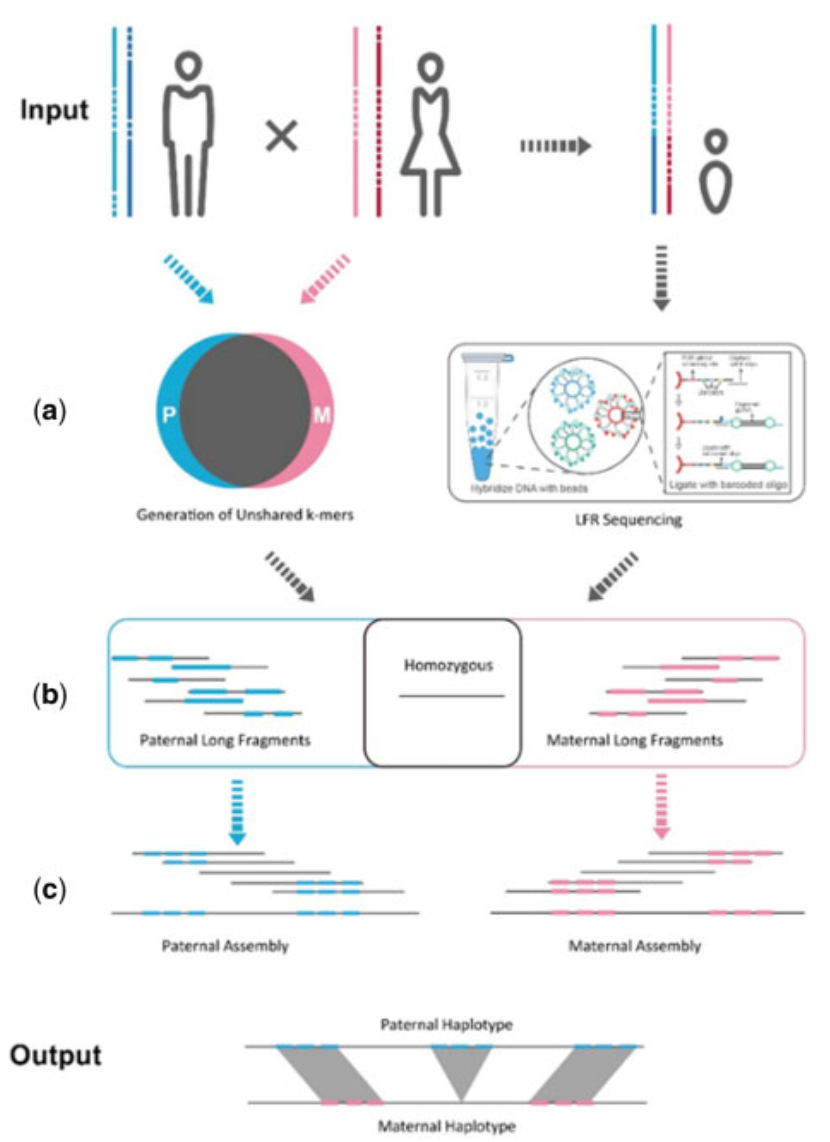
Understanding how genetic variations are inherited and structured within an individual’s genome is key to advancing precision medicine and evolutionary studies. In our latest publication in Bioinformatics, we present Haplotype-resolved Assembly for Synthetic long reads using a Trio-binning strategy (HAST)—an approach that enhances haplotype assembly accuracy using Long Fragment Read (LFR) sequencing in trio samples.
Key Findings
-
Improved Trio-Based Haplotype Assembly:
We developed HAST, a new computational pipeline that efficiently resolves haplotypes using trio-based sequencing data. By incorporating parental genomic information, HAST successfully phases long genomic sequences, improving both haplotype precision (~99.7%) and recall (~95.9%). -
High-Quality Genome Assembly Using LFR:
Building on prior advancements in LFR sequencing (originally introduced by Complete Genomics), we applied this technique to human trios. The combination of LFR sequencing and Hi-C data enabled chromosome-level genome assembly, facilitating more accurate reconstruction of both paternal and maternal haplotypes. -
Structural Variations and Single-Base Accuracy:
HAST demonstrated superior single-base accuracy (up to Q65) compared to other trio-binning methods, such as TrioCanu, while preserving structural variants (SVs) effectively. The method was particularly beneficial in detecting complex genomic rearrangements that traditional alignment-based haplotyping might miss.
Reflections
This study represents a significant milestone in our efforts to advance haplotype assembly using LFR sequencing in human trio datasets. Complete Genomics was among the earliest to introduce LFR technology, showcasing its role in haplotype phasing in previous Nature publications. The core idea behind LFR sequencing is similar to BAC library sequencing—effectively enabling sequence reconstruction across long genomic fragments, making it an ideal method for haplotype assembly.
In this research, we integrated multiple genomic datasets to refine haplotype reconstruction, ultimately generating haplotype-resolved assemblies that can serve as sequencing standards for evaluating genome assembly and analytical tools. While LFR sequencing has its challenges, such as read length limitations, our study illustrates its effectiveness in producing highly accurate haplotype-resolved assemblies. The successful completion of this work was made possible through the dedication of our team and collaborators.
The full text of this study can be accessed online at Bioinformatics.